«Volgiti ’n dietro e tien lo viso chiuso;
ch‚ se ’l Gorgón si mostra e tu ’l vedessi,
nulla sarebbe di tornar mai suso».
Tratto da Dante, “Divina Commedia”, Inferno, Canto IX
Quelle che verranno illustrate in questo articolo sono due condizioni che sfiorano (nonché sfidano) il confine tra la concretezza della scienza e il pàthos della mitologia. Anzi, per la precisione in queste malattie il mito diventa supplizio, poiché i sortilegi di Medusa agiscono con tormentante lentezza, senza che sia possibile l’intervento di un eroico Perseo, ma con la sola certezza di una fine estenuante.

Malattie pietrificanti
Si parla della fibrodisplasia ossificante progressiva (FOP) e dell’eteroplasia ossificante progressiva (PHO), patologie congenite caratterizzate dalla ingravescente ed inarrestabile trasformazione dei tessuti molli in osso. Due condizioni terribili, due vere situazioni di “malattia dell’uomo di pietra” (secondo nome della FOP) che, prevedibilmente, risultano molto rare. Difatti per quanto riguarda la FOP, se ne conta un caso ogni due milioni di persone (circa 2000-2500 persone affette in totale), senza sostanziali differenze in termini di età, sesso ed aree geografiche; per questa motivazione è difficile fare una stima precisa della situazione a livello mondiale ed italiano, per il quale panorama sembrano esserci trenta pazienti in totale. Ancora più infinitesimale è la situazione della PHO, patologia di recente definizione clinica (1994) per la quale si stimano circa sessanta casi in tutto il mondo.
Manifestazione ed evoluzione della FOP
La FOP, oltre ad essere letale nel suo decorso, risulta oltretutto subdola: difatti non è possibile porre rapidamente il sospetto della stessa (o comunque del fatto che ci sia qualcosa che non va) dato che alla nascita il bambino risulta in apparenza pressoché normodotato. Va detto che spesso è presente qualche anormalità congenita a livello dell’alluce di entrambi i piedi, soprattutto alluce valgo, malformazione del primo metatarso e/o monofalangismo; tuttavia queste anomalie non sono tipiche di questa sola malattia e talvolta costituiscono reperti isolati, quindi non possono essere prese come elemento diagnostico della patologia.
È verso i vent’anni (o anche prima, in certi casi) che iniziano a comparire delle tumefazioni, piccoli noduli di consistenza solida e caldi al tatto (a causa del processo infiammatorio in atto), rintracciabili a livello scapolare ed in prossimità del collo. I tessuti interessati sono principalmente muscoli, tendini, legamenti. Queste protuberanze sono dure, sempre di più col passare del tempo, fino ad assumere la struttura, la compattezza e la solidità proprie del tessuto osseo. La malattia però non evolve solo in senso qualitativo: i noduli tendono infatti ad aumentare progressivamente le loro dimensioni, ne nascono di nuovi e diffusi anche in corrispondenza del petto, dell’addome, del dorso, degli arti ed eventualmente anche del cranio; tuttavia non vengono interessati lingua, diaframma e sfinteri.
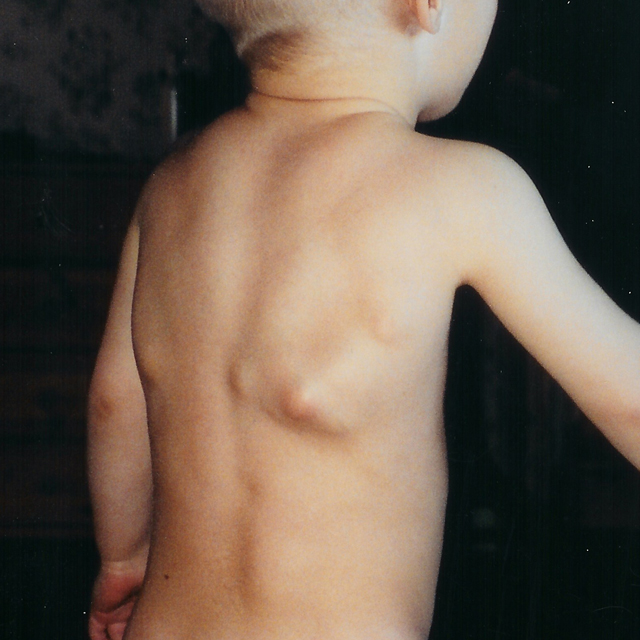
La velocità con cui progredisce la malattia e la gravità ad essa correlata sono molto variabili, ma con ogni probabilità anche semplici lesioni traumatiche e non (quali iniezioni intramuscolari, infezioni virali, stretching muscolare, cadute o affaticamento) possono portare a riacutizzazioni, peggioramenti e nascita di nuovi noduli. Questo fa sì che anche il tentativo di rimuovere chirurgicamente questi ponti ossei si riveli controproducente, poiché viene in tal modo scatenata un’ancora più violenta ed ingravescente ossificazione.
Una vera e propria trappola di cristallo (anzi, di pietra), i cui effetti sono a dir poco devastanti: a causa della trasformazione, i muscoli perdono infatti la loro funzione, si contraggono irregolarmente provocando spasmi e viene impedito il corretto movimento. Non solo, all’ossificazione tissutale si sovrappone la costruzione di un secondo scheletro esterno che, inglobando e facendo fondere al suo interno le articolazioni, nel lungo termine “ingabbia” il paziente senza che questo possa uscirne. Ne risulta gravemente compromessa la funzionalità cardiorespiratoria, fatto che porta ad una morte precocissima (l’età mediana è quarantuno anni). Spesso si associa anche una parziale o totale perdita dell’udito.
Il caso più famoso è quello di Harry Eastlack il cui corpo era talmente ossificato che poco prima della sua morte poteva solo muovere le labbra. Il suo scheletro è ora in mostra al Mutter Museum.

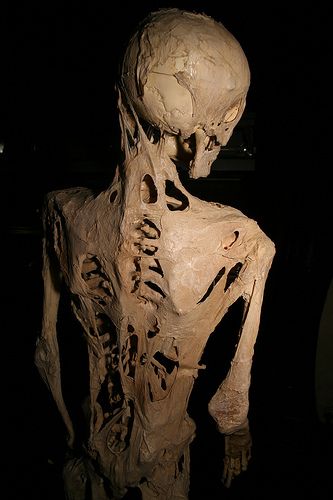
Sono stati descritti pazienti affetti da forme atipiche di FOP che:
- o presentano i segni classici della malattia associati ad un segno aggiuntivo o a diversi segni atipici (anemia aplastica concomitante, craniofaringioma, glaucoma infantile, ritardo della crescita), ed in questo caso si parla di FOP plus;
- o presentano significative variazioni di uno o di entrambi i segni classici della patologia (alluci normali o grave accorciamento delle dita), e queste forme costituiscono varianti della FOP.
Manifestazione ed evoluzione della PHO
La PHO presenta logicamente moltissime affinità con la FOP (nella sola ossificazione, poiché non sono generalmente presenti malformazioni ossee alla nascita), tuttavia l’insorgenza della malattia risulta molto più precoce, in quanto già nell’infanzia compaiono dei noduli a livello cutaneo. Tali rilevatezze, in maniera analoga a quanto detto nel precedente paragrafo, portano ad una progressiva trasformazione dei tessuti molli in osso, ma i primi tessuti ad esserne interessati non sono muscoli e fasce, bensì la stessa cute: si formano vere e proprie placche ossee aventi disposizione casuale, di dimensioni e collocazioni variabili. La malattia si estende poi allo stesso tempo verticalmente ed orizzontalmente, poiché col tempo le placche si approfondiscono a coinvolgere il tessuto sottocutaneo includendo eventualmente il tessuto muscolare, i tendini e legamenti, giungendo a queste strutture tramite i nervi e le pareti dei vasi sanguigni, a loro volta ossificati in una rete che ingabbia la muscolatura. Nel mentre si vengono a creare nuovi noduli cutanei ossificanti, aventi posizione casuale (a mosaico), o focalizzata in uno specifico distretto (sebbene gli arti siano le zone più comunemente affette, le ossificazioni possono anche coinvolgere il capo, il tronco, l’addome, la pelvi e il dorso). La velocità di progressione della malattia è variabile da soggetto a soggetto ed anche all’interno dello stesso paziente, ma non sono mai presenti riacutizzazioni improvvise. Le conseguenze sono naturalmente devastanti, soprattutto quando le placche insorgono in vicinanza delle articolazioni, le quali vengono inesorabilmente bloccate: più che letale, la malattia risulta gravemente invalidante. Comunque, dalle informazioni disponibili nei pochi adulti con PHO conosciuti, sembra che la malattia progredisca molto più lentamente nell’età adulta.
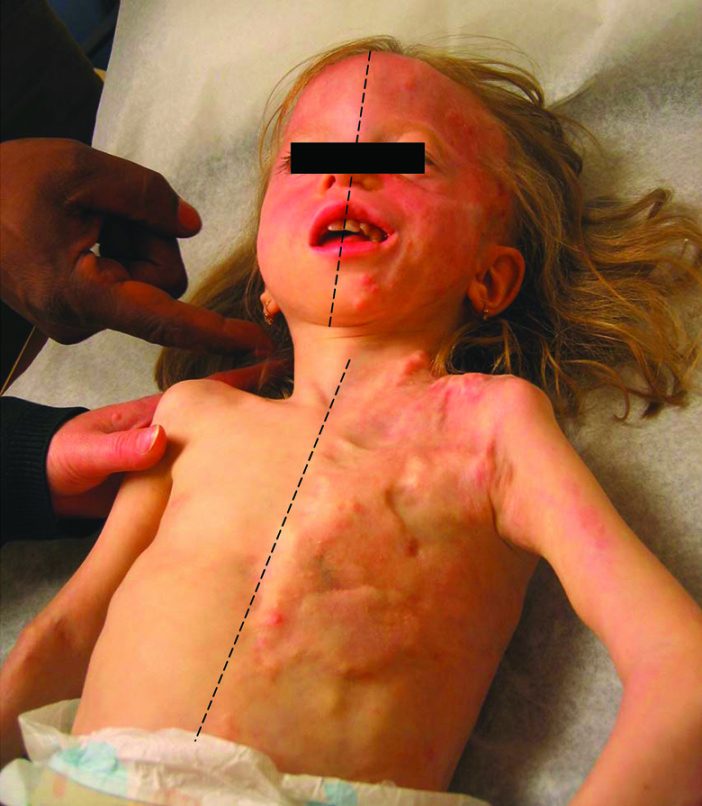 Esistono delle condizioni strettamente associate alla malattia:
Esistono delle condizioni strettamente associate alla malattia:
• l’Osteodistrofia Ereditaria di Albright (AHO) è una rara condizione genetica in cui si formano nella cute e nel tessuto sottocutaneo piccole quantità di ossificazioni in associazione con malformazioni scheletriche minori e una alterata risposta delle ghiandole endocrine ad alcuni ormoni (riguarda soprattutto le paratiroidi);
• l’Osteoma Cutis, forma minore della PHO caratterizzata dalla mancanza della progressione delle ossificazioni verso i tessuti profondi, la quale limita il problema alla sola pelle.
Le cause delle patologie
Alla base di questi quadri tanto complessi quanto disarmanti ci sono mutazioni geniche specifiche, trasmissibili da genitore a figlio per via autosomica dominante (il che implica 50% di probabilità che un figlio erediti la mutazione e manifesti la malattia), o nate come mutazione spontanea durante la vita intrauterina.
Per quanto riguarda la FOP, la mutazione incriminata è stata scoperta nel 2006. Come riportato da diversi e recenti lavori di ricerca, questa malattia è scatenata da una mutazione della proteina AVCR1, codificata da alcuni geni presenti sul cromosoma 2. Questo tipo di proteina appartiene alla categoria delle BMP, costituita da potenti regolatori della formazione ossea, tanto che questi fattori sono attualmente usati nella pratica ospedaliera nell’ambito della riparazione delle fratture; peraltro, nella loro espressione fisiologica tali proteine hanno anche un ruolo di protezione in relazione ai processi di fibrosi che si determinano nel corso delle infiammazioni. È perciò una proteina di fondamentale importanza per la regolazione ed il mantenimento dell’omeostasi (ovvero dell’auto-conservazione dell’organismo al mutare delle condizioni esterne dell’ambiente). Nel corso della FOB, dunque, il loro ruolo viene amplificato in misura abnorme, al punto da far perdere la loro funzione di controllo e dare inizio alla cosiddetta ossificazione eterotopica; è questo processo che, come detto in precedenza, porta ad una crescente ed aberrante trasformazione del corpo.
Tale tipologia di ossificazione interessa anche la PHO, ma in questo caso alla base della malattia c’è una mutazione della proteina GNAS, codificata da alcuni geni sul cromosoma 20. Non è del tutto chiaro come ciò possa dare luogo all’ossificazione eterotopica, dato che tale proteina è presente su tutte le cellule: l’ipotesi più accreditata afferma che ad essere coinvolte siano le cellule staminali dei tessuti sopra citati, le quali perderebbero un importante fattore di regolazione e tenderebbero a divenire cellule del tessuto osseo. Per entrambe le patologie, comunque, molti aspetti rimangono oscuri, e lasciano letteralmente di sasso.
Diagnosi delle malattie di pietra
La sola clinica non è sufficiente per poter porre diagnosi di queste rare malattie ossee, a meno che le stesse non siano già ad uno stadio molto avanzato: certamente non deve essere trascurato il dettaglio delle malformazioni congenite degli alluci spesso presenti nella FOP, ma come già accennato questo dettaglio può soltanto dare un primario orientamento. Nella PHO la clinica ha un suo iniziale (ma limitato) ruolo diagnostico solo quando compaiono francamente le placche ossee.
Con la radiologia si riesce ad avere più informazioni relativamente alla condizione di muscoli e tendini, informazioni che poi la TC e la RM descrivono più nel dettaglio. A volte potrebbe essere necessario eseguire biopsie per escludere altre condizioni, ma le stesse spesso non vengono effettuate se c’è il sospetto della malattia per evitare il rischio di riacutizzazioni (anche la POH, pur non presentando riacutizzazioni, vede un certo peggioramento nel suo decorso dopo l’attuazione della biopsia). La diagnosi definitiva viene posta tramite analisi genetica, tuttavia (complice il fatto che ancora non esiste un test prenatale) spesso l’esito è tardivo.
Terapia e gestione dei pazienti affetti
Come già detto, non c’è ancora un Perseo che possa sconfiggere questa potente Medusa; ma, sempre rimanendo nella metafora di questo mito, non dev’essere dimenticato che, nonostante le varie differenze nelle versioni della storia, tra le tre sorelle Gorgoni l’unica mortale è Medusa. È quindi un bersaglio che si può potenzialmente sconfiggere una volta individuatone i punti deboli.
Proprio in questo risiede l’importanza della ricerca medica, paradossalmente poco finanziata in questo ambito (data la rarità delle due malattie), ma comunque molto attiva.
Malgrado ciò, allo stato attuale non esistono terapie efficaci: il trapianto di midollo osseo si è rivelato controproducente.
Quello che si può fare è cercare di gestire le condizioni in cui il paziente si trova a vivere, in modo da evitare urti improvvisi (deleteri per la FOP), prevenire sovra infezioni, e trattare il dolore (frequente soprattutto nelle fasi terminali e nelle riacutizzazioni) attraverso l’utilizzo di varie classi di farmaci analgesici ed eventualmente con la radioterapia. Importante infine la gestione umana: spesso questi pazienti rischiano la solitudine, tendono ad isolarsi e vengono isolati a causa degli handicap provocati dalla loro condizione (i malati di FOP, ad esempio, nell’ultimo decennio della loro breve vita sono costretti alla sedia a rotelle). Oltre alla vicinanza dei familiari, sono provvidenziali in tal senso le associazioni che si occupano specificatamente della cura di questi pazienti, in modo che gli stessi abbiano un’esistenza il più possibile serena.